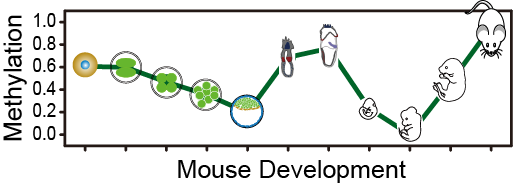
Mouse Developmental Methylome Database
Home
MethyBrowser
Download
Submit
Tutorial
About Us
Links
MethyBrowser
Recommended Browser: Mozilla Firefox (1024*768)