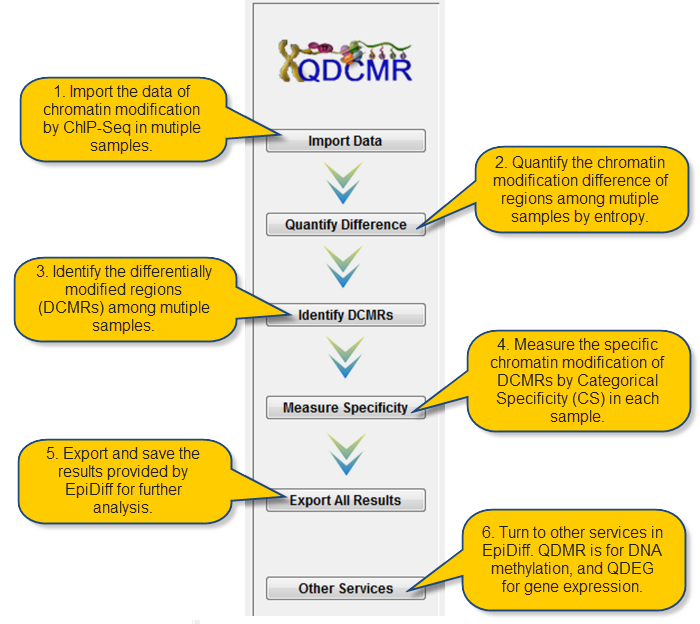
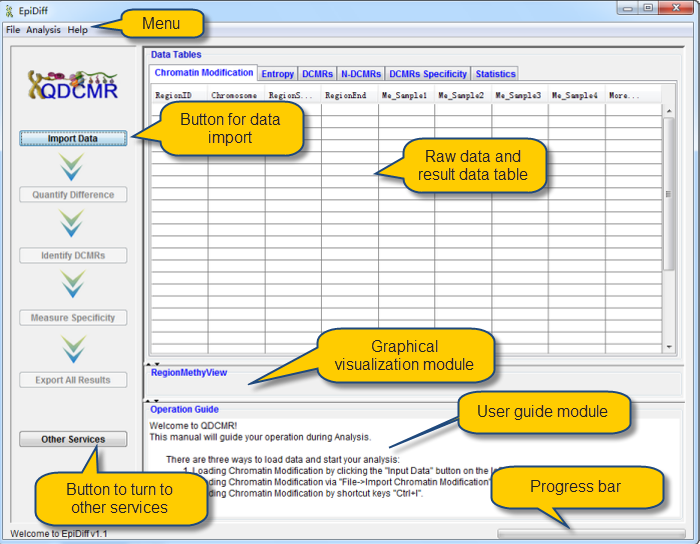
There are three ways to load data and start your analysis:
| |
1) |
Loading chromatin modification data by clicking the "Import Data" button on the left panel. |
| |
2) |
Loading chromatin modification data via "File->Import chromatin modification Data". |
| |
3) |
Loading chromatin modification data by shortcut keys "Ctrl+I". |
QDCMR provides the visual interface to import data by which users can import two types of data.
One is the data processed by users in which each region have chromatin modification levels in each samples.
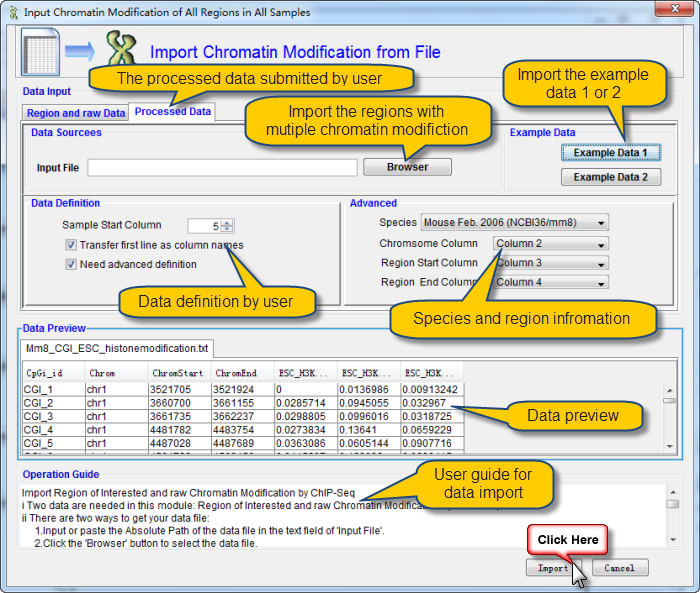
i There are two ways to get your data file:
1.Input or paste the Absolute Path of the data file in the text field of "Input File".
2.Click the "Browser" button to select the data file.
ii Important Notices:
1.It is suggested to refer the example data by click the "example Data 1 or 2" button before import your own data.
2.Information about the regions of interest should be before the chromatin modification data for the region.
3.You can tune the start column of chromatin modification data.
4.Select the Check Box named "Transfer first row as column names" if you want column names as your first row.
5.Column names will be generated automatically if you don't check the box named "Transfer first row as column names".
6.You must ensure that there are no missing values in your data.
7.The first 20 rows of data will be shown in the data file preview window as any change take places.
Before import data, please make sure that the data is save in txt or xls file as the format as shown in this example data:
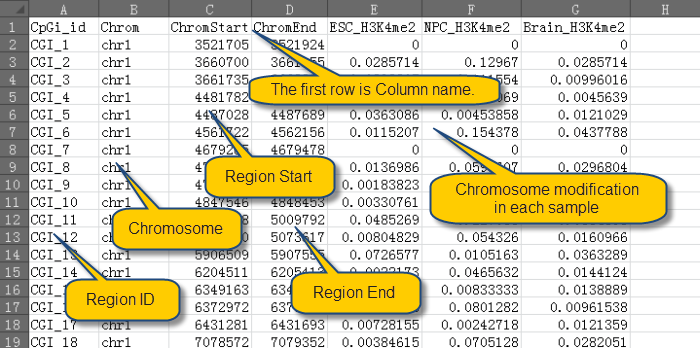
Now Import chromatin modification Data from Txt or Xls File:
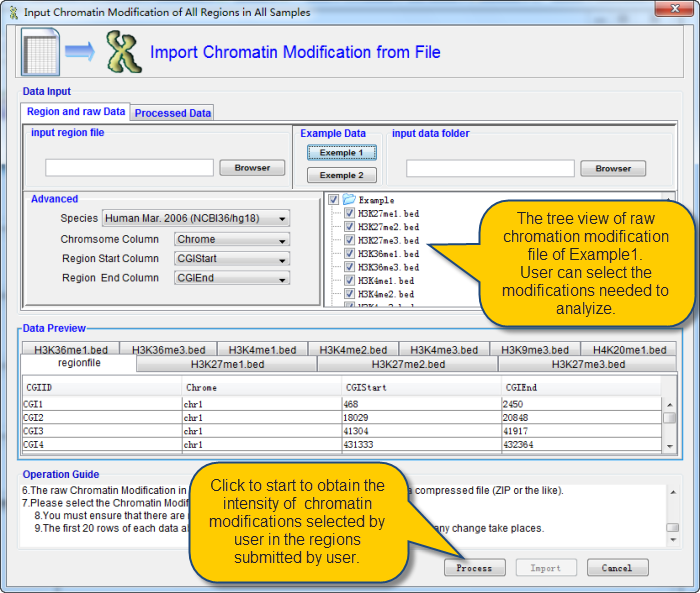
Another one is the raw ChIP-Seq data by the panel "Region and raw data". The details of input parameters as show following:
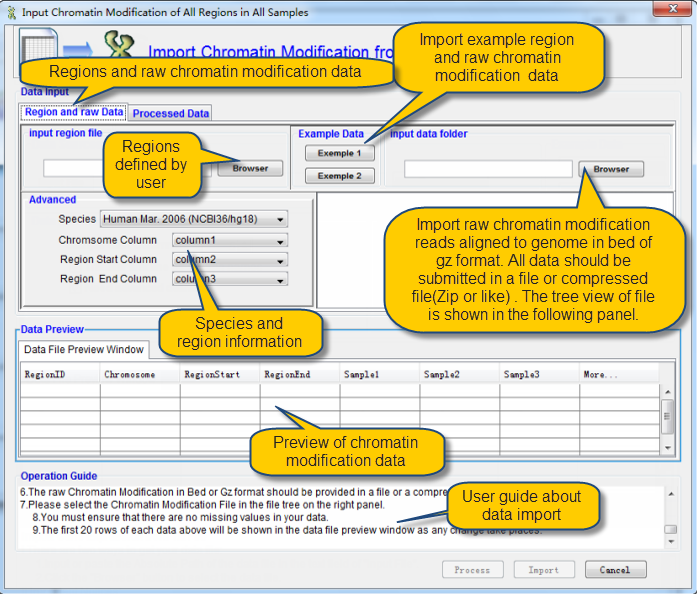
Before import data, please make sure that the data is save in the right format. The region file should be saved as txt or xls file as the format as shown in this example data:
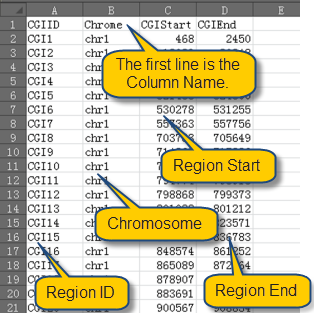
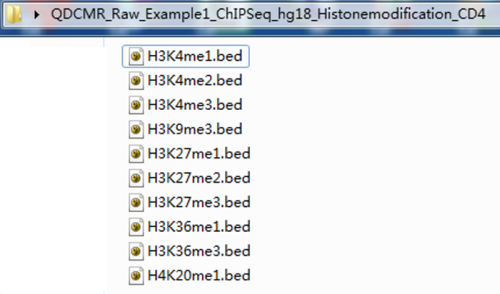
And the raw files including chromatin modification reads which has been aligned to genome, can be save in bed format, and slao can be txt.gz format which is widely used in ChIP-Seq data. What's more, these files can be put in a file folder or a compressed file (Zip or like). The examples for this two format can be found in following.
The first exmaple is for the bed files in a file folder.
The second exmaple is for the txt.gz files in a compressed file (.zip).
Now Import chromatin modification Data.
Take the Example1 as example. User can selcet the files he wants to analysize. Then click the button "Process" on the bottom.
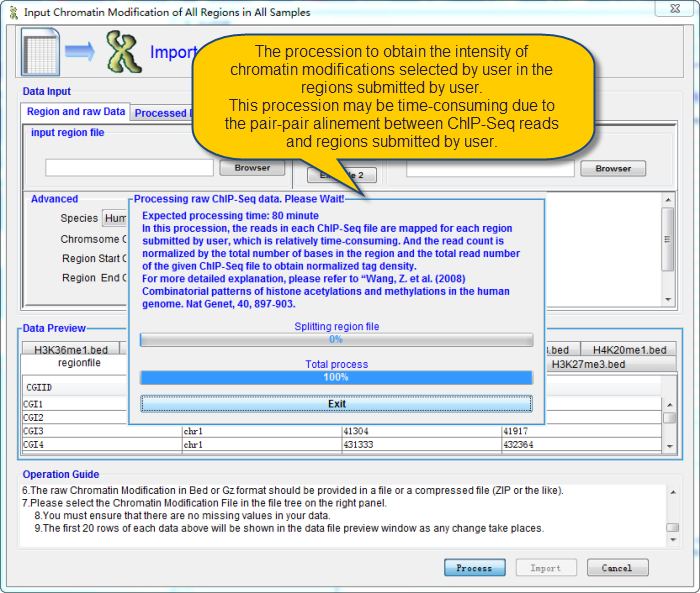
QDCMR will do the alinement between the regions submitted by user and the chromatin modification reads. This progess may be time-consuming due to the pair-pair alinement between ChIP-Seq reads and regions submitted by user.
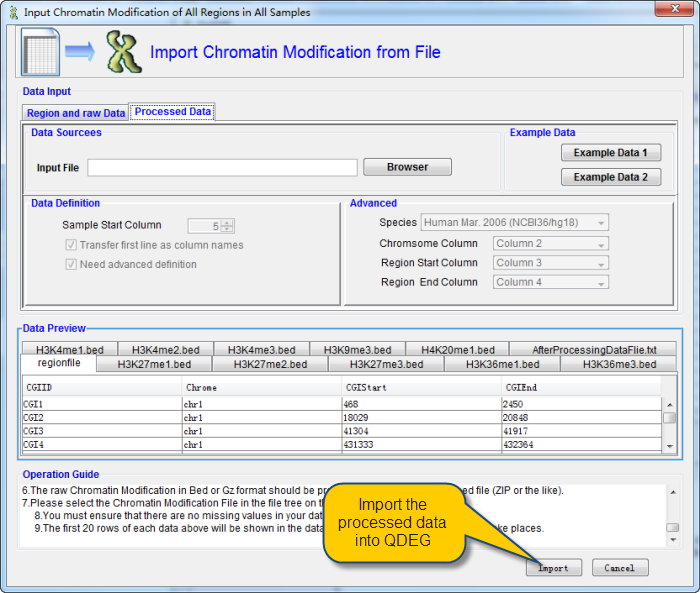
Import Region of Interested and raw Chromatin Modification by ChIP-Seq
i Two data are needed in this module: Region of Interested and raw Chromatin Modification by ChIP-Seq.
ii There are two ways to get your data file:
1.Input or paste the Absolute Path of the data file in the text field of "Input File".
2.Click the "Browser" button to select the data file.
iii Important Notices:
1.It is suggested to refer the example data by click the "example Data 1 or 2" button before import your own data.
2.Information about the regions of interest can be defined by users.
3.Please make sure that the first row of region file is column names.
4.Please select the species you need and the corresponding columns for chromosome, region start, and region end.
5.The raw Chromatin Modification used in current version of QDCMR in only that produced by ChIP-Seq.
6.The raw Chromatin Modification in Bed or Gz format should be provided in a file or a compressed file (ZIP or the like).
7.Please select the Chromatin Modification File in the file tree on the right panel.
8.You must ensure that there are no missing values in your data.
9.The first 20 rows of each data above will be shown in the data file preview window as any change take places.
When the alinement is completed, the software will turn to the interface as following:
After confirmation, click Import button to import data. The following interface will be shown a little while.
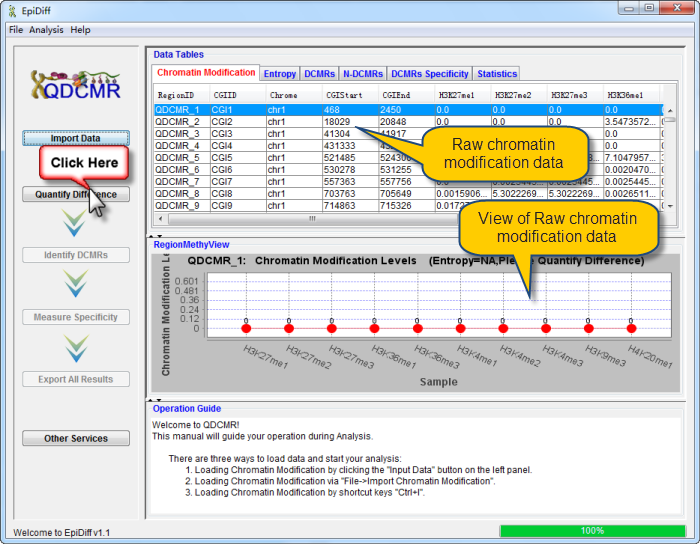
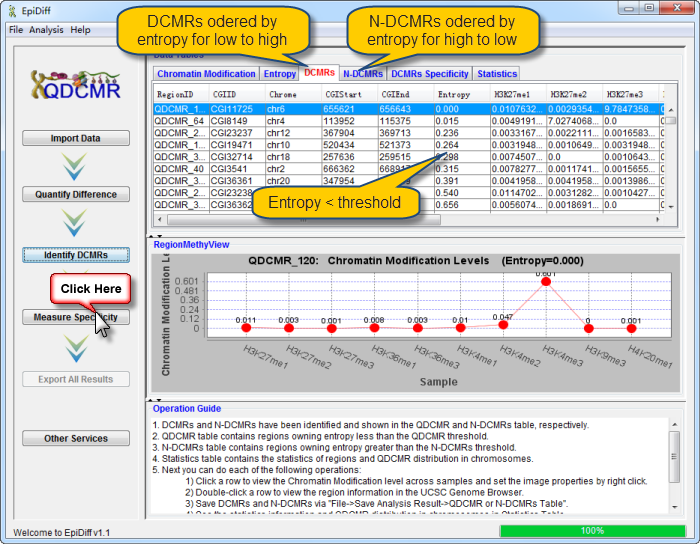
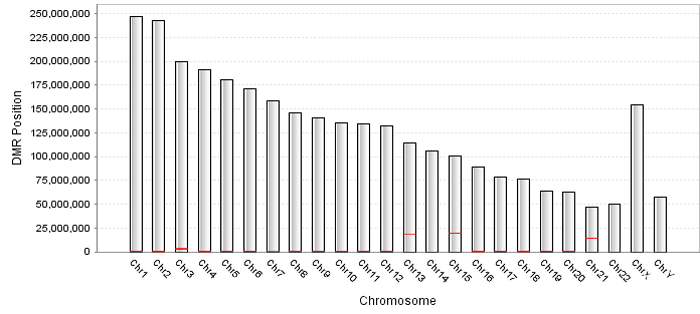
Chromatin modification data has been successfully loaded and shown in the data table. The first column named "RegionID" is generated automatically as the ID for each region in QDCMR.
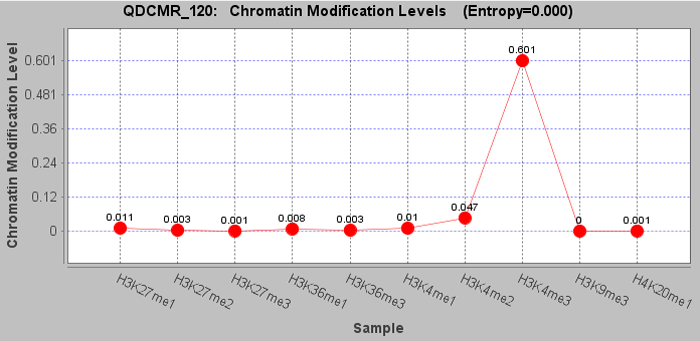
chromatin modification levels in the first row were shown in RegionMethyView acquiescently.
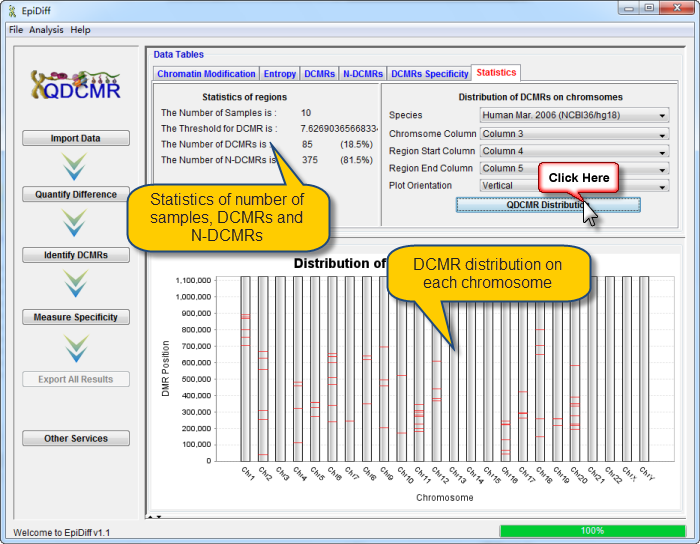
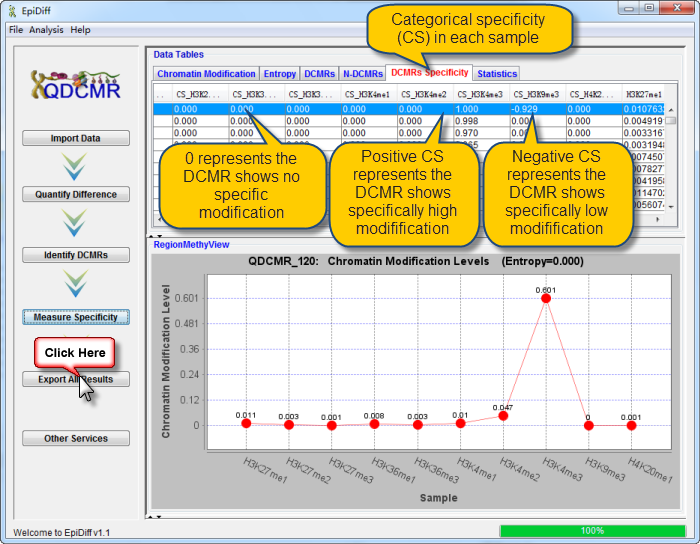
Next you can do each of the following operations:
| |
1) |
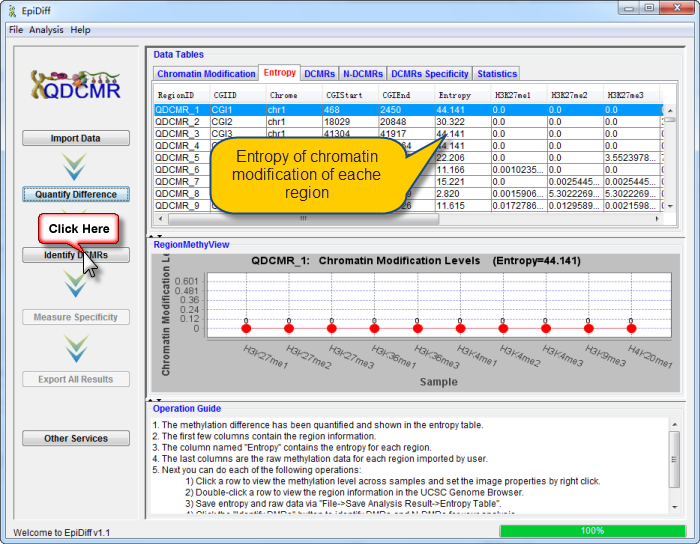
Click a row to view the chromatin modification level across samples and set the image properties by right click. |
| |
2) |
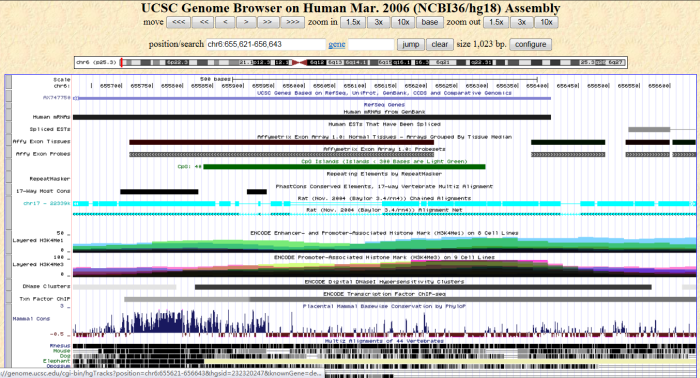
Double-click a row to view the region information in the UCSC Genome Browser. |
| |
3) |
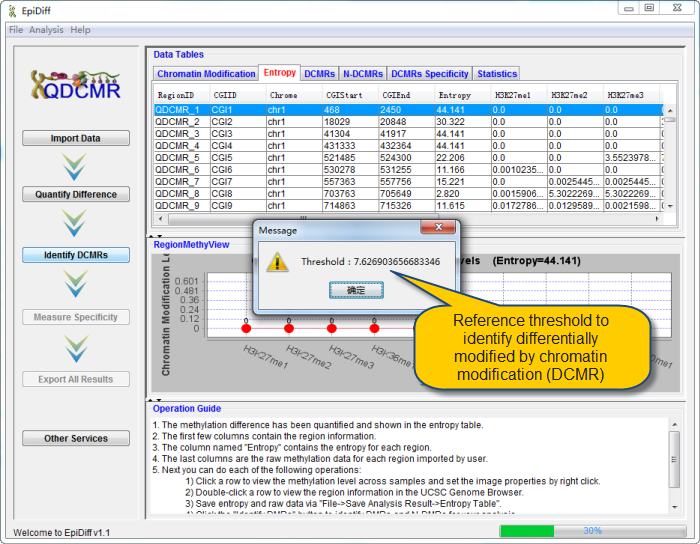
Click "Quantify Difference" button to quantify chromatin modification difference by calculating entropy for all regions. |
|