|
Tutorials and FAQ about QDEG |
 |
|
|
 |
^Top |
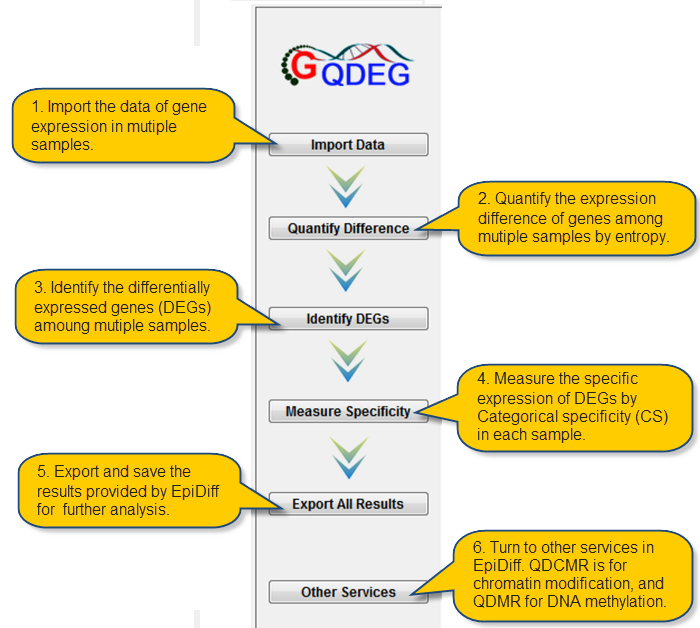 |
 |
^Top |
QDEG, as the third module in EpiDiff, provides a quantitative approach to quantify gene expression difference and identify differentially expressed genes (DEGs) from genome-wide gene expression profiles by adapting Shannon entropy. Its platform-free and species-free nature makes it easy for computational biologists to analysis the gene expression and epigenetic regulation across various temporal and spatial gene expression profiles.
This Tutorial contains the following sections:
| Getting Started |
Describes how to start Local and online QDEG. |
| Import Data |
Describes how to preprocess and import gene expression data. |
| Quantify Difference |
Describes how to quantify gene expressio difference across various samples. |
| Identify DEGs |
Describes how to identify DEGs by threshold imbedded in QDEG. |
| Measure Specificity |
Describes how to measure sample-specificity for each DEGs. |
| Export Results |
Describes how to save results. |
| Visualization |
Describes how to display gene expression level, DEG distribution and UCSC links. |
|
| Getting Started |
^Tutorial |
^Top |
| This section describes how to start online and local QDEG.
Before start QDEG, user should start EpiDiff using two ways:
1. Start EpiDiff directly via Java Web Start, using the Java Web Start feature (advantage: no installation required);
2. Download EpiDiff installation package, install and run it locally on your machine (advantage: no network required).
And then, click one the button "Other Services" to change to QDEG service in the following picture.
When first started, QDEG displays the home page.
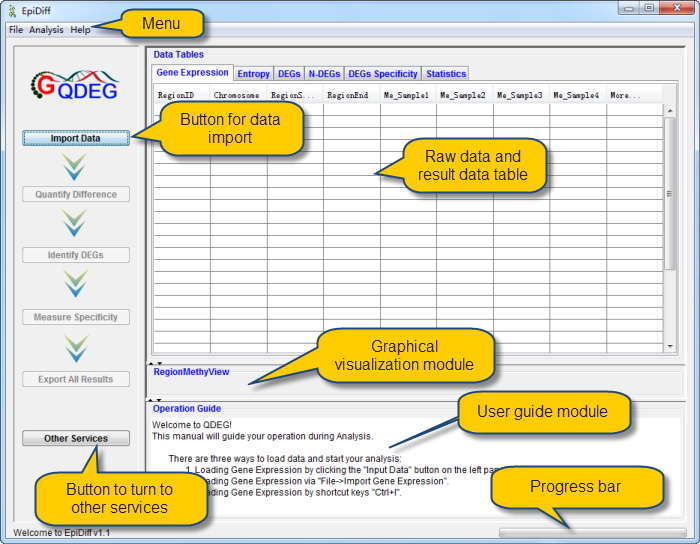 |
| Import Data |
^Tutorial |
^Top |
There are three ways to load data and start your analysis:
| |
1) |
Loading Gene expression data by clicking the "Import Data" button on the left panel. |
| |
2) |
Loading Gene expression data via "File->Import Methylation Data". |
| |
3) |
LoadingGene expression data by shortcut keys "Ctrl+I". |
QDEG provides the visual interface to import data.
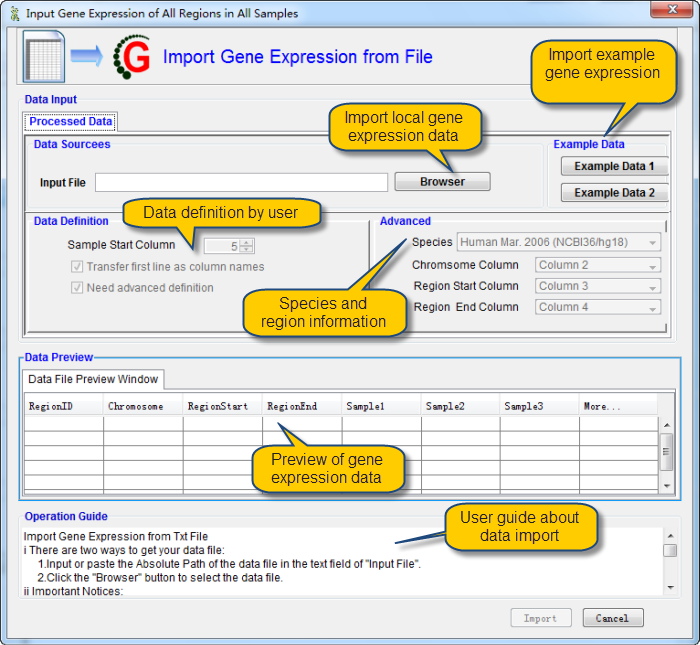
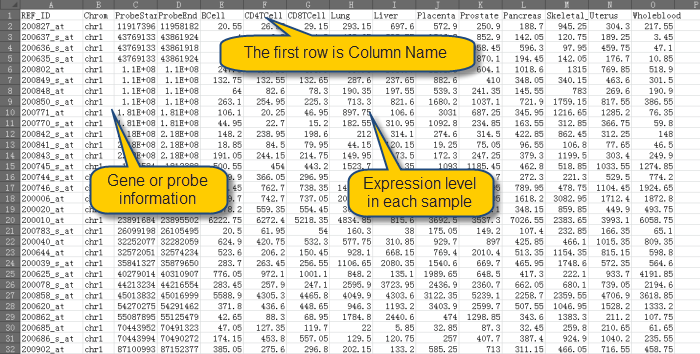
Before import data, please make sure that the data is save in txt or xls file as the format as shown in this example data:
Now Import Methylation Data from Txt or Xls File:
i There are two ways to get your data file:
1.Input or paste the Absolute Path of the data file in the text field of "Input File".
2.Click the "Browser" button to select the data file.
ii Important Notices:
1.It is suggested to refer the example data by click the "example Data 1 or 2" button before import your own data.
2.Information about the regions of interest should be before the methylation data for the region.
3.You can tune the start column of methylation data.
4.Select the Check Box named "Transfer first row as column names" if you want column names as your first row.
5.Column names will be generated automatically if you don't check the box named "Transfer first row as column names".
6.You must ensure that there are no missing values in your data.
7.The first 20 rows of data will be shown in the data file preview window as any change take places.
After confirmation, click Import button to import data. The following interface will be shown a little while.
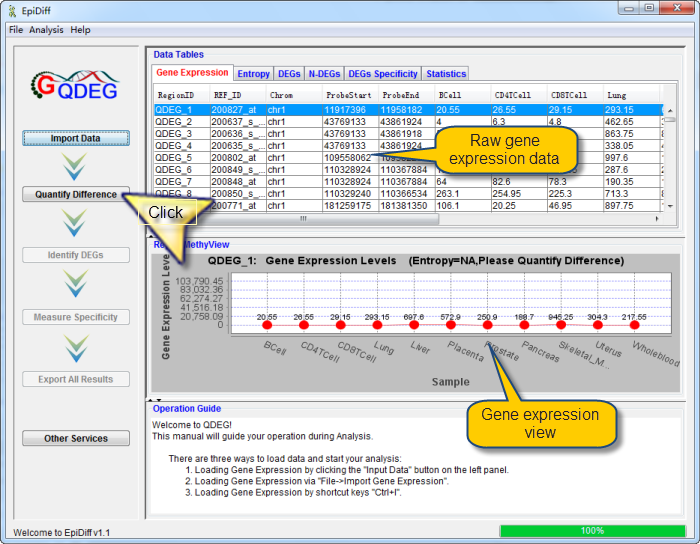
Gene expression data has been successfully loaded and shown in the data table. The first column named "RegionID" is generated automatically as the ID for each region in QDEG.
Gene expression levels in the first row were shown in RegionMethyView acquiescently.
Next you can do each of the following operations:
| |
1) |
Click a row to view the gene expression level across samples and set the image properties by right click. |
| |
2) |
Double-click a row to view the region information in the UCSC Genome Browser. |
| |
3) |
Click "Quantify Difference" button to quantify gene expression difference by calculating entropy for all regions. |
|
| Quantify Difference |
^Tutorial |
^Top |
Click "Quantify Difference" button to quantify gene expression difference by calculating entropy for all regions.
The following interface about quantified gene expression difference will be shown when the progress bar reaches 100%.
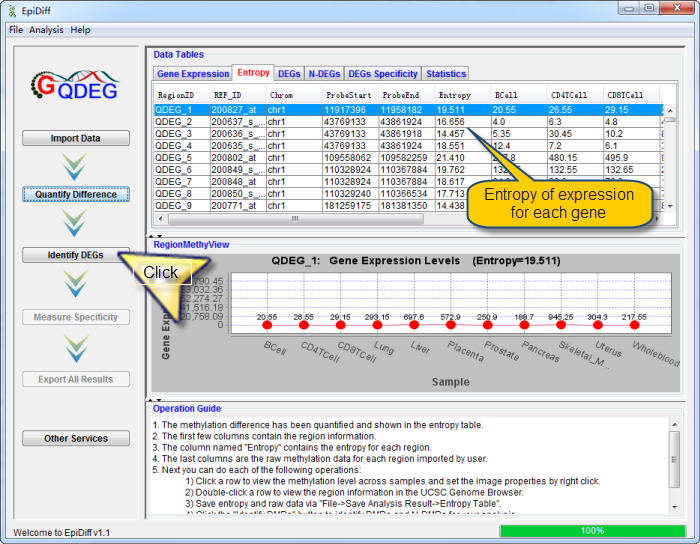
The gene expression difference has been quantified and shown in the entropy table. The first few columns contain the region information. The column named "Entropy" contains the entropy for each region. The last columns are the raw gene expression data for each region imported by user.
You can do each of the following operations:
| |
1) |
Click a row to view the gene expression level across samples and set the image properties by right click. |
| |
2) |
Double-click a row to view the gene information in the UCSC Genome Browser. |
| |
3) |
Save entropy and raw data via "File->Save Analysis Result->Entropy Table". |
| |
4) |
Click the "Identify DEGs" button to identify DEGs and N-DEGs for your analysis. |
|
| Identify DEGs |
^Tutorial |
^Top |
| Click the "Identify DEGs" button to identify DEGs and N-DEGs for your analysis.
The interface shows the reference threshold for identification of DEGs will be provided.
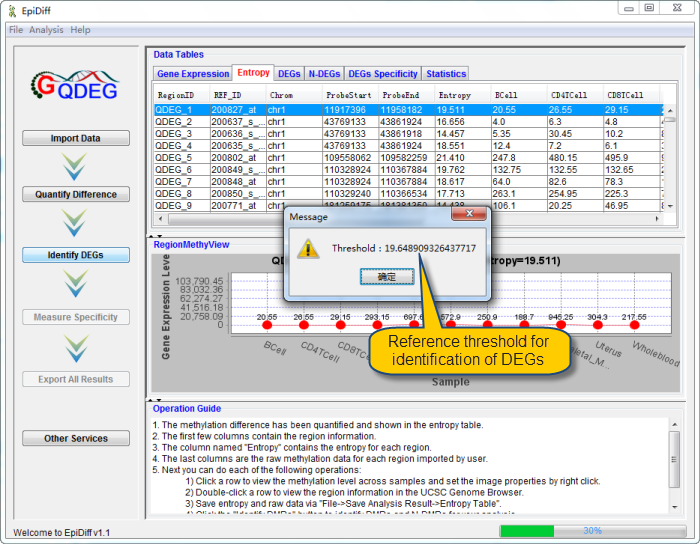
QDEG will identify DEGs from imported regions by the threshold defined by user. The following interface about DEGs will be shown when the progress bar reaches 100%.
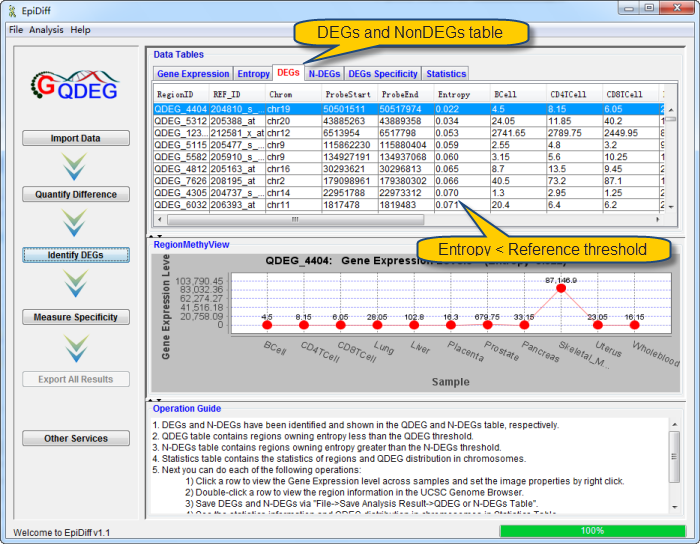
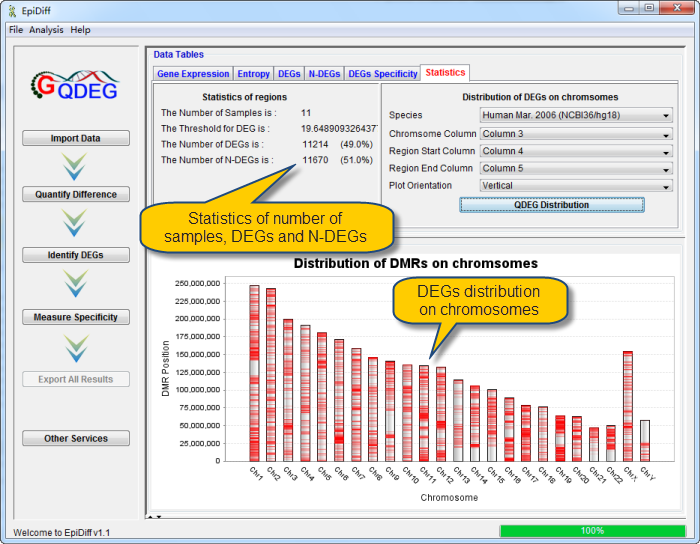
DEGs and N-DEGs have been identified and shown in the DEG and N-DEG table, respectively. DEG table contains regions owning entropy less than the DEG threshold. N-DEG table contains regions owning entropy greater than the DEG threshold.. Statistics table contains the statistics of regions. And DEG distribution in chromosomes can be shown by clicking the button "QDEG Distribution".
Next you can do each of the following operations:
| |
1) |
Click a row to view the expression level across samples and set the image properties by right click. |
| |
2) |
Double-click a row to view the region information in the UCSC Genome Browser. |
| |
3) |
Save DEGs and N-DEGs via "File->Save Analysis Result->DEG or N-DEG Table". |
| |
4) |
See the statistics information and DEG distribution in chromosomes in Statistics Table. |
| |
5) |
Click the "Measure Specificity" button to measure the sample specificity for all DEGs. |
|
| Measure Specificity |
^Tutorial |
^Top |
| Click the "Measure Specificity" button to measure the sample specificity for all DEGs. QDEG will calculate the categorical specificity for each region in each sample. The following interface about sample-specificity of each DEG will be shown when the progress bar reaches 100%.
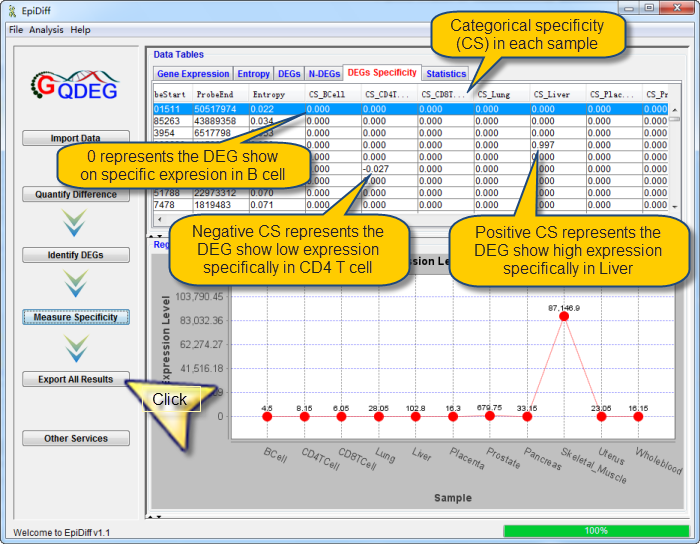
The measurement of sample specificity has been finished and shown in the Specificity Table. The first few columns contain the region information. The column named "Entropy" contains the entropy for each region. The columns named as "CS_" contain the specificity of each region in every sample. The last columns are the raw methylation data for each region imported by user.
Next you can do each of the following operations:
| |
1) |
Click a row to view the methylation level across samples and set the image properties by right click. |
| |
2) |
Double-click a row to view the region information in the UCSC Genome Browser. |
| |
3) |
Save Specificity Table via "File->Save Analysis Result->Specificity Table". |
| |
4) |
Save All Results by clicking "Export All Results" or via "File->Save Analysis Result->All Results". |
|
| Export Results |
^Tutorial |
^Top |
Save All Results by clicking "Export All Results" or via "File->Save Analysis Result->All Results". The results will be saved in a file containing Entropy Table, DEG Table, N-DEG Table, Specificity Table and Statistics. And then you can analysis the DEGs and N-DEGs in the biological process in which you are interested.
|
| Visualization |
^Tutorial |
^Top |
| First, in order to increase the visual effect of methylation data, QDEG provides visualization module GeneExpView. GeneExpView shows the levels across various samples and DEG distribution on each chromosome. User can reset and save the figure by right clicking.
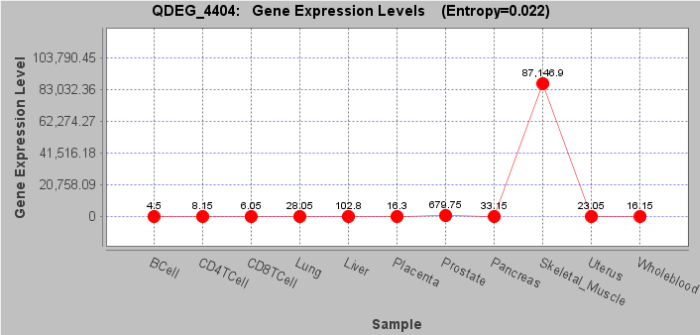
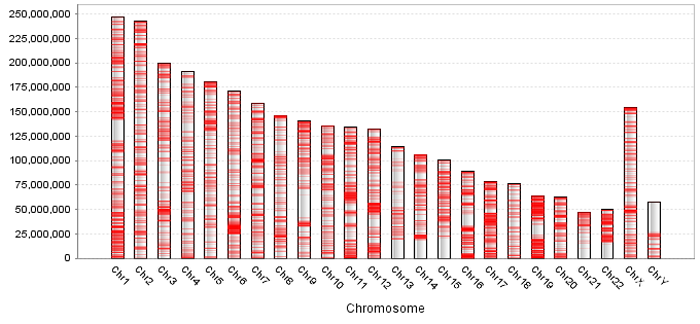
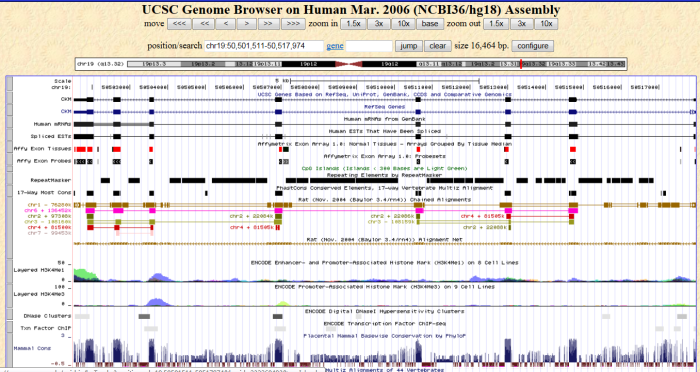
Second, in order to show the information near each gene in user's data, QDEG provides the link to UCSC Genome Browser. User can view the genome features near each gene, such as transcript, miRNA, SNP, GC percent, CpG island, and histone modifications et al. This view will facilitate the study between DEGs and other regulatory elements in the genome.
|
 |
^Top |
1. Public Server/General
| |
1) |
Where can I find the hardware and software prerequisites for QDEG? |
| |
A. |
The hardware requirements, supported operating systems, and supported browsers is shown in Download site. |
| |
2) |
How can I get help with QDEG or provide feedback? |
| |
A. |
The tutorial fully describes QDEG and how to use it.
To provide feedback or ask a question please do not hesitate to contact Hongbo Liu (hongbo919@gmail.com) or Yan Zhang (yanyou1225@yahoo.com.cn). |
2. Useage
| |
1) |
How can I find QDEG in EpiDiff? |
| |
A. |
QDEG is embedded in EpiDiff as the second module. QDEG service can be found as Tutorial about getting started. |
| |
2) |
How the threshold of DEGs in QDEG is made? |
| |
A. |
QDEG is a quantitative approach to quantify gene expression difference and identify DEGs from genome-wide gene expression profiles by adapting Shannon entropy. For more details can refer to a previous paper by us listed following.
Based on the quantified gene expression difference among multiple sample, the thresholds for identification of DEGs are obtained as Schug et al. did in selecting tissue-specifically expressed genes from gene expression profiles [1]. Here, the log base 2 of the fold change between replicate-dependent difference from the average level across replicates and the theoretical maximum range of gene expression was assumed as a normal distribution with mean equal to zero and a standard deviation (s). In the example above, setting s =0.25, the mean expression intensity was sampled from the distribution of observed mean gene expression intensities, and 5000 regions with uniform gene expression across 11 samples were modeled. Then the sampled entropy values were calculated by QDEG. These 5000 entropy values follows a normal distribution in which a threshold was determined at p = 0.05 (one-sided). This process was repeated 10 times, and the mean value of 10 thresholds defined as the reference threshold for identification of DEGs. The similar discription can be found in our QDMR paper [2].
[1]Schug J, Schuller WP, Kappen C, Salbaum JM, Bucan M, Stoeckert CJ, Jr.: Promoter features related to tissue specificity as measured by Shannon entropy. Genome Biol 2005, 6:R33. [Abstract] [PDF]
[2]
Zhang Y, Liu H, Lv J, Xiao X, Zhu J, Liu X, Su J, Li X, Wu Q, Wang F, Cui Y: QDMR: a quantitative method for identification of differentially methylated regions by entropy. Nucleic Acids Res 2011, 39:e58. [Abstract] [PDF] |
3.Data Formats
| |
1) |
What file format dose QDEG support? |
| |
A. |
Currently, QDEG can supports Txt or Xls file. In the latter version, more file formats will be supported. |
| |
2) |
Where can I find information about file formats used by QDEG? |
| |
A. |
Information on file formats supported by the modules currently in QDEG is available in Tutorial. |
4.Other
If you have any trouble or recommendations, please do not hesitate to contact Hongbo Liu (hongbo919@gmail.com) or Yan Zhang (yanyou1225@yahoo.com.cn).
|
|
CopyRight © Group of
Computational Epigenetic Research
College of Bioinformatics
Science and Technology, Harbin Medical University, China
|
|
Recommended Browser: Mozilla Firefox
(1024*768)
|
|
|
|
|



